This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is a protein?
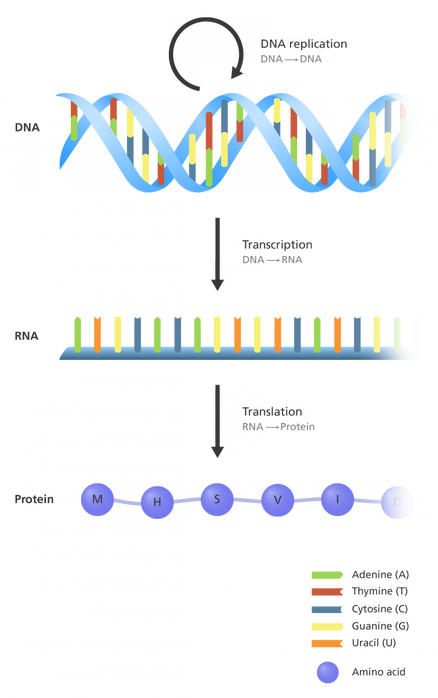
If you have experience in a field other than science, you may have heard the word 'protein' in your day to day life without realizing the importance of proteins in your body's functions. You would not be alive without proteins, in fact you wouldn't even have developed!
Your genome is several feet of DNA compacted in the nucleus of each one of your cells. The DNA has a long sequence of four nucleotides, A, C, T and G. The sequence of these acids in your genome is "read" and transcribed into RNA, which is the messenger that translates the information stored in your genome into functional proteins. Each set of three nucleotides (A, C, T or G) has a specific combination that calls for one subunit of a protein, called an amino acid.
These amino acids string together in the order that your RNA tells them to. Once this string of amino acids becomes long enough, it folds into its functional structure.
The functional structure of this string of amino acids is called a protein. Proteins are integral parts of everything your body does, including your allergic reactions!
Your genome is several feet of DNA compacted in the nucleus of each one of your cells. The DNA has a long sequence of four nucleotides, A, C, T and G. The sequence of these acids in your genome is "read" and transcribed into RNA, which is the messenger that translates the information stored in your genome into functional proteins. Each set of three nucleotides (A, C, T or G) has a specific combination that calls for one subunit of a protein, called an amino acid.
These amino acids string together in the order that your RNA tells them to. Once this string of amino acids becomes long enough, it folds into its functional structure.
The functional structure of this string of amino acids is called a protein. Proteins are integral parts of everything your body does, including your allergic reactions!
What is Protein Phylogeny?
There is a relationship between all living organisms. This relationship lies within our DNA, and phylogenetics is the way we quantify that relationship. Phylogenetics is based on mathematical probability. It's a way for us to analyze the relationships of species and genes over evolutionary history.
Recent advancements in the methods scientists use to sequence genes have made the study of phylogenetics even more powerful, because we can track changes down to the specific nucleotide. In protein phylogenomics, we are interested in protein sequences rather than gene sequences. The major mechanisms we use to build a tree include parsimony, likelihood, distance, and Bayesian methods.3 |
How are trees made?
Parsimony: Simplicity.
The tree with the fewest number of changes, or the most simple tree, is the best.
Likelihood: Probability.
This deals with the likelihood of a change to occur in the sequence, based on biological principles.
Distance: Variation.
The length of the branches of a tree can mean many things, or nothing at all. When they follow the distance method, they account for the actual genetic distance of the sequences between species.
Bayesian Methods: Statistics.
This is the probability of the credibility of the tree based on the information acquired.
Following this set of ideas for building a tree can help us determine how the gene we want to study changed over time and how it is related across species.
You may be wondering how a similar gene can exist in several species. This is the definition of an ortholog. An ortholog is a pair of similar genes that exist in the genomes of different species. This means that two species can share essentially the same gene! As you can imagine, it is useful to study orthologs because of the physiological differences between species. The protein or gene in question may affect different species in unique ways! There are other ways that genes can travel along the evolutionary timeline, but this is the one we want to focus on when creating a tree.
The tree with the fewest number of changes, or the most simple tree, is the best.
Likelihood: Probability.
This deals with the likelihood of a change to occur in the sequence, based on biological principles.
Distance: Variation.
The length of the branches of a tree can mean many things, or nothing at all. When they follow the distance method, they account for the actual genetic distance of the sequences between species.
Bayesian Methods: Statistics.
This is the probability of the credibility of the tree based on the information acquired.
Following this set of ideas for building a tree can help us determine how the gene we want to study changed over time and how it is related across species.
You may be wondering how a similar gene can exist in several species. This is the definition of an ortholog. An ortholog is a pair of similar genes that exist in the genomes of different species. This means that two species can share essentially the same gene! As you can imagine, it is useful to study orthologs because of the physiological differences between species. The protein or gene in question may affect different species in unique ways! There are other ways that genes can travel along the evolutionary timeline, but this is the one we want to focus on when creating a tree.
Definitions
Neighbor Joining: Bottom up method for building a tree based on combining 'neighbors', or organisms sharing a recent node, into groups. [4]
Average Distance: Refers to the average genetic distance, or similarity, between two species' protein sequence.
BLOSUM62: Alignment method based on the probability of each nucleotide substitution as evolution occurs across species.
PiD: Percent identity. It's an alignment method based on perfect amino acid matches and internal gaps.5
Average Distance: Refers to the average genetic distance, or similarity, between two species' protein sequence.
BLOSUM62: Alignment method based on the probability of each nucleotide substitution as evolution occurs across species.
PiD: Percent identity. It's an alignment method based on perfect amino acid matches and internal gaps.5
Phylogenetic Trees for TGFB1
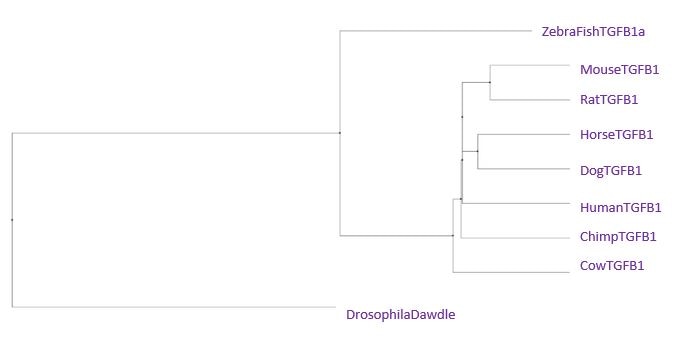
Neighbor Joining Tree using BLOSUM62
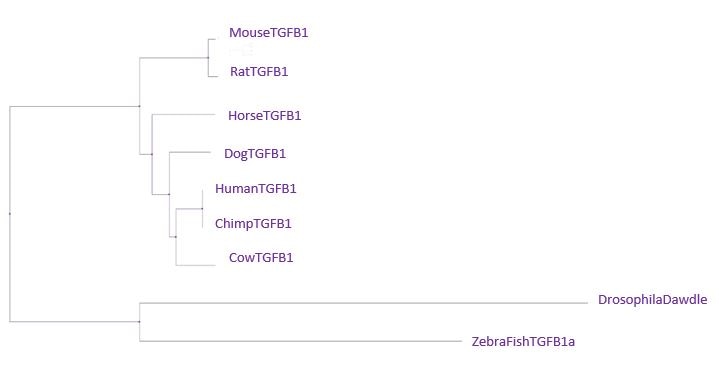
Neighbor Joining Tree using PiD
Average Distance Tree using BLOSUM62
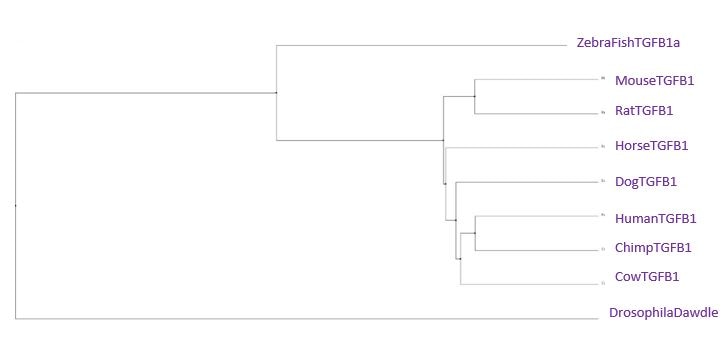
Average Distance Tree using PiD
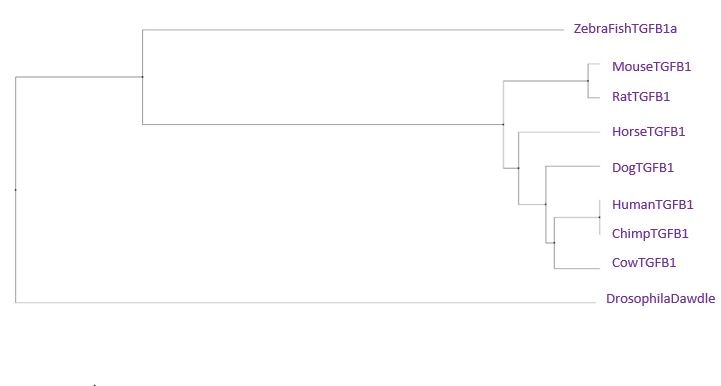
Phylogenetic Tree via Clustal Omega
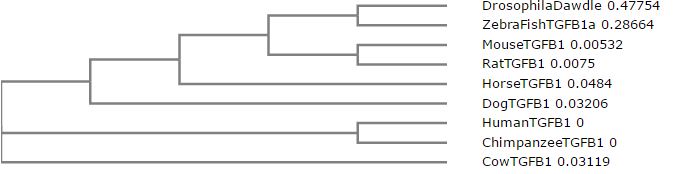
What do those numbers mean?
The numbers after the name of the species and the name of it's ortholog to the TGFB1 protein is a measure of distance. Between sequences, there is a distance of relatedness. If we think of the human TGFB1 protein as our baseline, then we can infer that the human TGFB1 protein has zero difference to our baseline. The numbers greater than zero next to the other species relate to how much they differ with our baseline.
Analysis
Overall, the most important feature of all of the trees is that the chimpanzee sequence is the same or extremely similar to the human sequence. That means that there was actually very little difference between our TGFB1 and a chimp's. The second interesting feature between the trees is the distance between the drosophila and the zebra fish. Usually, we would expect these species to be close together on the tree. The only tree we really observe this in is the Neighbor Joining Tree using PiD. Drosophila has a fairly unique homolog of the TGFB1 gene, which is most similar in amino acids and gaps to the zebra fish gene. Since the neighbor joining tree using percent identity joins based on a most recent common ancestor in combination with perfect amino acid matches and gaps, it makes sense the pair would be clustered together in this tree. The most important thing to remember is that although the TGFB1 protein structure is highly conserved across all of these species, the functionality can differ greatly based on the different ways each species uses the protein.
References
[1] Analysis Tool Web Services from the EMBL-EBI. (2013)McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez RNucleic acids research 2013 Jul;41(Web Server issue):W597-600 doi:10.1093/nar/gkt376
[2] Yang, K., & Zhang, L. (2008). Performance comparison between k-tuple distance and four model-based distances in phylogenetic tree reconstruction. Nucleic Acids Research, 36(5), e33. http://doi.org/10.1093/nar/gkn075
[3] Yang Z., Rannala B. (2012). Molecular phylogenetics: principles and practice. Nat Rev Renet, 13(5): 303-14. doi:10.1038/nrg3186
[4] Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. [PubMed]
[5] MegAlign Help. (n.d.). Retrieved April 30, 2017, from https://www.dnastar.com/megalign_help/index.html#!Documents/calculationofpercentidentity.htm
Images
[1] Edited from: https://viettes.wordpress.com/tag/oak-pollen/
[2] https://universe-review.ca/F10-multicell03.htm
[3] http://news.stanford.edu/news/multi/features/darwin/
[4] http://www.yourgenome.org/facts/what-is-the-central-dogma
[2] Yang, K., & Zhang, L. (2008). Performance comparison between k-tuple distance and four model-based distances in phylogenetic tree reconstruction. Nucleic Acids Research, 36(5), e33. http://doi.org/10.1093/nar/gkn075
[3] Yang Z., Rannala B. (2012). Molecular phylogenetics: principles and practice. Nat Rev Renet, 13(5): 303-14. doi:10.1038/nrg3186
[4] Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. [PubMed]
[5] MegAlign Help. (n.d.). Retrieved April 30, 2017, from https://www.dnastar.com/megalign_help/index.html#!Documents/calculationofpercentidentity.htm
Images
[1] Edited from: https://viettes.wordpress.com/tag/oak-pollen/
[2] https://universe-review.ca/F10-multicell03.htm
[3] http://news.stanford.edu/news/multi/features/darwin/
[4] http://www.yourgenome.org/facts/what-is-the-central-dogma
